Extraction and separation of fats and lipids
|
Principle |
|

|
The aim of all extraction procedures is to separate cellular or
fluid lipids from the other constituents, proteins, polysaccharides, small
molecules (amino acids, sugars...) but also to preserve these lipids for
further analyses.
There is a great diversity of methodologies
because biological tissues are not similar when considering their structure,
texture, sensitivities and lipid contents. The ideal solvent for lipid
extraction would completely extract all the lipid components from a sample,
while leaving all the other components behind. In practice, the efficiency of
solvent extraction depends on the polarity of the lipids present compared to
that of the solvent.
Polar lipids (such as glycolipids or phospholipids) are more soluble in polar
solvents (such as alcohols), than in non-polar solvents (such as hexane). On
the other hand, non-polar lipids (such as triacylglycerols) are more soluble in non-polar
solvents than in polar ones. The fact that different lipids have different
polarities means that it is impossible to select a single organic solvent to
extract them all. Thus the total lipid content determined by solvent extraction
depends on the nature of the organic solvent used to carry out the extraction:
the total lipid content determined using one solvent may be different from that
determined using another solvent.
Ethyl ether and petroleum ether are the
most commonly used solvents, but pentane and hexane are also used for some
foods.
|
Folch method |
|
|
Various solvents or solvent combinations have been suggested as
extractants, but most lipid analysts use chloroform-methanol (2:1 by volume) as
suggested by Folch.The extract is shaken and equilibrated with one fourth its
volume of a saline solution, when the mixture partitions into two layers, of
which the lower is composed of chloroform-methanol-water in the proportions
86:14:1 (by volume) and contains virtually all of the lipids, while the upper
phase consists of the same solvents in the proportions of 3:48:47 (by volume),
respectively, and contains much of the non-lipid contaminants. It is not always
recognised how important it is that the proportions of chloroform, methanol and
water in the combined phases should be as close as possible to 8:4:3 (by
volume), otherwise selective losses of lipids may occur. If carried out by the
book, this method can give reliable results.
- Homogenize the tissue with chloroform:methanol (2:1) to a
final dilution 20 times the volume of the tissue sample, i.e. the homogenate
from 1 g of tissue is diluted to a volume of 20 ml. The time of homogenization
will vary with the sample but a minimum of 3 minutes is usually required.
- Filter the homogenate through a suitable paper into a
glass-stoppered bottle. (Centrifugation may be used instead of filtration). For
the purpose of computation, this extract corresponds to 0.05 times its volume
of tissue, i.e. 1 ml of extract corresponds to 0.05 g of tissue.
- Wash the crude extract with 0.2 of its volume of either water
or salt solution.
- Allow the solution to separate into two phases. The volumes
of the upper and lower phases are 40 and 60% of the total volume
respectively.
- Remove the upper layer by siphoning.
- Rinse the interface three times with pure 'upper phase', i.e.
the chloroform:methanol:water 3:48:47 so that the lower phase is not disturbed.
This has the effect of removing any 'fluff' at the interface.
- Finally add methanol so that the lower phase and the rinsing
liquid form one phase.
- Dilute the resulting solution to any desired volume by the
addition of chloroform:methanol (2:1).Steps 7 and 8 may be omitted if it is
intended to remove the solvent under vacuum to yield a dry extract for
weighing.
|
Bligh & Dyer method |
|
|
The Bligh and Dyer method is a simple adaptation of this method
and was developed merely as an economical means of extracting lipids from
tissues such as fish muscle, which contain relatively little lipid and a high
proportion of water. Bligh and Dyer do clearly state that for quantitative
extraction of lipids, it is necessary to perform a re-extraction of the tissue
residue with chloroform alone and add this extract to the filtrate prior to
evaporation of the solvent. This would improve the yield of non-polar
lipid.
Procedure:
- To a sample containing 1 ml water, add 3.75 ml of a mixture
chloroform/methanol (1/2)
- Vortex during 10-15 min
- Add 1.25 ml chloroform with mixing 1 min and 1.25 ml water
with mixing another minute before centrifugation.
- Discard the upper phase and collect the lower phase through
the protein disk with a Pasteur pipette For large volumes of liquid, it is
advisable to filter the mixture to remove the insoluble parts of the sample and
to centrifuge the liquid phase to allow the formation of the two liquid
phases.
- After evaporation, the lipid extract (lower phase) will be
redissolved in a small volume of chloroform/methanol (2/1).
This basic procedure was improved to increase the yield of
lipids. One of the most common modifications is to replace water by 1M NaCl.
This addition blocked the binding of some acidic lipids to denatured lipids. If
necessary, the addition of 0.2 M phosphoric acid to the salt solution is
possible (Hajra, lipids, 1974, 9, 502) to improve their recovery. In this case,
plasmalogens are converted to lyso lipids. If an exhaustive extraction is
necessary, an extraction with two steps can be used.
|
Liquid-solid extractions |
|
|
The term "solid-phase" or "sorbent
extraction", frequently abbreviated to "SPE", simply implies a physical
extraction process involving a liquid and a solid phase. In practice, it has
come to mean the use of commercial pre-packed columns containing stationary
phases related to those used widely in high-performance liquid chromatography
(HPLC), that may be adsorbents such as silica gel, reversed-phase materials or
ion-exchange media. The packing material is held in a place within a plastic
column by porous frits, also constructed of a plastic material, and the column
ends in a Luer tip to facilitate connection to a vacuum manifold, to a needle
or to a collection vessel.
The objective of the analyst is ideally to
isolate a component of interest from a more complex sample in a pure
concentrated state. This might be achieved by choosing conditions so that the
required analyte is retained on the column while the impurities pass straight
through, or conversely by allowing the analyte to elute through while the
impurities are retained. In some applications of SPE columns in lipid analysis,
this ideal objective can indeed be attained. On the other hand, many lipids are
rather similar in their physical properties and it may only be practicable to
isolate groups of lipid classes of related polarity.
|
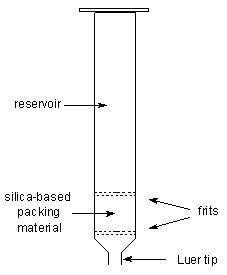 |
| A solid-phase extraction column |
|
Specific applications of SPE columns published to date in lipid
analytical methodology are described in the excellent pages of the
Lipid Analysis Unit at the Scottish Crop Research
Institute.
|
Thin-layer chromatography |
|
|
|
Thin-layer chromatography consists of a stationary phase
immobilized on a glass or plastic plate and a solvent. The sample, either
liquid or dissolved in a volatile solvent, is deposited as a spot on the
stationary phase. The constituents of a sample can be identified by
simultaneously running standards with the unknown. One edge of the plate is
then placed in a solvent reservoir and the solvent moves up the plate by
capillary action. When the solvent front reaches the other edge of the
stationary phase, the plate is removed from the solvent reservoir. The
separated spots are visualized with ultraviolet light or by placing the plate
in iodine vapor. The different components in the mixture move up the plate at
different rates due to differences in their partioning behavior between the
mobile liquid phase and the stationary phase. |
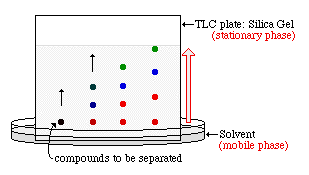 |
| Scheme of a Thin Layer Chromatography |
|
More information on chromatographic methods are presented in the
pages of
Science Hypermedia.
|